Nat Mach Intell |深度学习识别单克隆细胞系

原创 xiaoyudian 图灵基因 今天
收录于话题 前沿生物大数据分析
撰文:xiaoyudian
Nature子刊
推荐度:⭐⭐⭐⭐⭐
亮点:
1、文中形成了一个框架,在这个框架中,具有模块化设计的深度学习算法可以自动验证明场显微镜中的单克隆性,所需的标记相对较少。
2、文中将工作流程的功能进一步扩展到菌落形态分类,展示了在自动化工作流程中自主监测单克隆细胞系发育和克隆选择的潜力。
2021年5月7日,美国纽约干细胞基金会研究所的研究人员Brodie Fischbacher等人合作在《Nature Machine Intelligence》上发表了一篇“Modular deep learning enables automated identification of monoclonal cell lines”的文章。文中报告设计了一个深度学习工作流,自动检测菌落的存在,并从细胞成像识别克隆性。该工作流程被称为Monoqlo,它集成了多个卷积神经网络,关键的是,利用了细胞培养过程的时间方向性。我们的算法设计提供了一个完全可扩展的,高度可解释的框架,能够在一个小时内分析工业数据量使用商品硬件。我们关注的是人类诱导的多能干细胞的单克隆化,但我们的方法是可推广的。Monoqlo标准化了单克隆过程,使菌落选择协议可以无限扩大,同时最小化技术可变性。


从培养群体中提取的单细胞的分离和随后的扩增建立单克隆性,这通常被认为是培养高质量细胞系的必要步骤。这一步骤旨在减少或消除基因组和表型的异质性,试图最大限度地提高细胞系的均匀性。人类诱导多能干细胞是单克隆化关键的细胞培养过程(iPSCs)。它为体外模拟疾病状态提供了巨大的希望,使非侵入性遗传关联研究成为可能。由于这一步骤在诱导多能干细胞的自动化和高通量衍生过程中一直是一个关键的瓶颈,因此我们将此细胞类型作为研究单克隆化方法的案例。
目前验证单克隆性的唯一方法是人工检查显微镜成像,分选后定期进行菌落生长跟踪。基于卷积神经网络(CNNs)的深度学习已经应用于干细胞研究的许多过程,包括自动推断分化和预测IPSC衍生细胞的功能。但目前CNNs从未被用于自动识别任何细胞类型的单克隆化协议的克隆性。
在本文中,研究者报告了一种算法设计,通过利用细胞培养过程固有的时间方向性来克服这些困难。我们的计算工作流,称为Monoqlo,集成了多个CNNs,每个都有自己的模块化功能。Monoqlo提供了一个高度可伸缩的框架,能够在一小时内使用商用硬件分析数以万计的图像数量的数据集。通过将自动干细胞培养和深度学习相结合,这项工作展示了机器学习应用于从明场显微镜鉴定单克隆细胞系的一个例子。
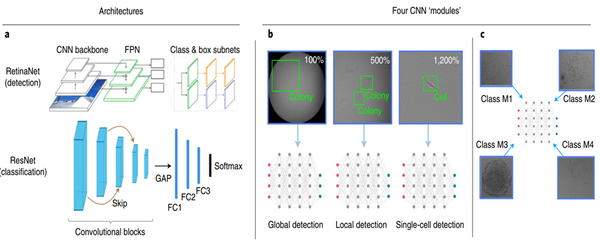
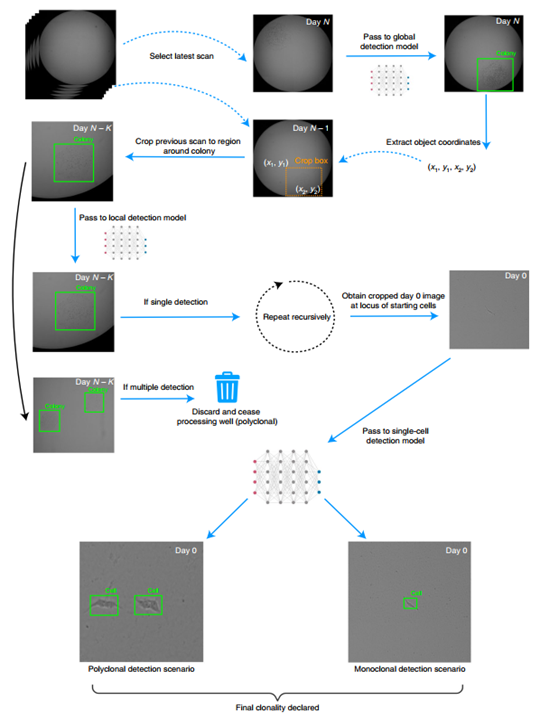
首先将自动分配克隆性的任务模块化为四个不同的深度学习功能(图1)。模块化的决定是基于初步调查期间作出的经验推断。单一模型不能很好地处理单克隆化过程中使用的图像放大倍数和对象类别的多样性。因此,研究中根据时间、放大倍数和作物级别对训练集进行分层,训练四个单独的神经网络,每个都有自己的模块功能。首先,将全局检测用于检测完整图像中菌落的存在与否。其次,在不同的变焦放大倍数下,在不同的裁剪图像中检测菌落的任务称为局部检测。第三,把在完全放大的裁剪图像中的单个细胞任务称为单细胞检测。最后通过使用视网膜网络检测体系结构来完成上述三个任务,实现形态学分类。

本研究中设计了Monoqlo计算工作流来整合训练过的神经网络。该算法以逆时间顺序处理基本图像。在实际的例子中,这是一个经过裁剪的图像,只是为了删除图像的黑色边框,保留了物理的整个领域。这些图像被传递到全局检测模型,其输出是一个坐标向量,用于划分任何被检测菌落的边界框。然后,该算法将这些坐标展开,加载下一个最近的图像,并将图像裁剪到结果区域。然后,生成的图像被传递给局部检测模型,该模型报告早期群体的边界框,在与裁剪坐标求和时表示其在原始未裁剪图像中的位置。在此过程中,工作流程的这种增量、迭代处理以及裁切盒尺寸的扩展是必不可少的,因为由于非径向增长和扫描之间的微小位置变化,每天都会有与精确同心度的小偏差。经过几天的成像,这些偏差加起来会形成相当大的偏移量。因此,简单地在后期菌落的中心位置进行裁剪和放大,很难得到一个或多个起始细胞所在的视场。
除了计数单个起始细胞外,如果观察到两个或更多明显不同的细胞团,假定它们来源于来自相同FACS类型的两个或更多细胞,通常可以推断出多克隆性。如果全局或局部检测模型在按时间顺序向后迭代的过程中任何一点报告了群集计数>1,算法相应地声明是多克隆的,并停止对任何图像的进一步处理。或者,如果工作流程继续准确地检测一个菌落,直到到达第0天扫描,产生的图像将被放大并精确地裁剪在一个或多个祖先细胞周围。然后可以将此图像传递给单细胞检测模型,提供起始细胞数量的计数。在此基础上,该孔最终可能被宣布为单克隆或多克隆。
接下来评估学习轨迹,并对每个CNN在各自任务中的预测性能进行基准测试。在目标检测网络的情况下,研究中的初始评估指标是在一个保留的验证数据集上测试损失函数值的变化,该数据集代表了最初分配的图像集的20%。重要的是,来自原始培训或验证集的任何数据都没有用于整个Monoqlo框架的下游验证。本研究手动评估模型的性能,通过视觉比较标签和预测的验证图像与各自的边界框绘制。从这些比较中,我们根据两个指标来量化检测性能:被正确预测和分类的标记对象的百分比和假阳性的数量,其中模型检测到一个不存在的对象,作为分析的图像总数的比率。最后,生物学家使用图像比尺测量的真实菌落宽度,通过monoqlo预测的边界盒X维。

最后对Monoqlo作为一个统一的、模块化的工作流程的有效性进行了基准测试,首先在一个手动管理的、类平衡的校验集上测试其准确性,然后在一个原始的、未经过滤的、来自真实世界的单克隆运行数据集上评估其克隆识别性能。我们精心设计的测试集包括从人工分类的历史记录中随机选择的空、单克隆和多克隆三类的100个孔。每个孔开始处理的成像日期从8天到18天随机生成。实际场景验证是在单克隆化作业(DMR0001)中进行的,共包含768个孔,时间跨度为19天,因此产生了18,240张图像。人工检查图像发现其中561个是空的。因此,Monoqlo识别出了194个非空的孔,包括115个单克隆情况,其中发现有2个和5个孔分别有多克隆和空的真实分类,剩余的108个(93.9%)与真实情况相符合。最后,61个孔是多克隆的,其中57个孔是真实的,4个孔是单克隆。
综上所述,本研究提出了使用深度学习对象检测方法自动识别克隆性。首先基于深度学习构建了Monoqlo的框架,允许从原始数据集中通过算法分割和裁剪菌落,还可以自动过滤出空孔的图像,这些图像通常代表了图像的绝大部分。在许多情况下,研究人员也可以根据他们对给定种群或孔的最新图像进行的分类,对图像进行批处理。应用Monoqlo可以追溯地将诸如是否分化的标签分配给种群的早期实例。这可以减少对未经过滤的图像集进行大量费力的手工检查和标记的需要,使未来的模型能够部分或完全自主地生成大量的训练量。因此,我们的算法为生成定制数据集提供了一个宝贵的工具,用于未来深入学习在iPSC研究中的效用调查。
教授介绍


Daniel Paull,美国纽约干细胞基金会自动化系统和干细胞生物学副总裁。Daniel Paull帮助开发了防止线粒体疾病遗传的方法,研究核转移,并参与了广泛的其他干细胞相关研究。同时,在干细胞研究和高通量自动化的交叉领域,他领导着一个由生物学家、工程师和数据科学家组成的团队,致力于进一步加深我们对人类生物学的理解,推动新疗法的发展。目前,Daniel Paull博士在Nature、Cell Stem Cell等杂志发表上多篇文章。
参考文献
Fischbacher, B., Hedaya,S., Hartley, B.J. et al. Modular deep learning enables automated identificationof monoclonal cell lines. Nat Mach Intell(2021).https://doi.org/10.1038/s42256-021-00354-7.