[E::faidx_adjust_position] The sequence not found; samtools/bcftools mpileup problem #1015
Comments
|
In fasta, anything following a space is comment, so the sequence name is |
|
My case. I took random data from SRA for training purposes. Then I got SAM and FASTQ via SRA Tools and converted SAM to BAM using SAMtools. The first 10 lines of FASTQ. The first 10 lines of SAM. I tried to get BCF with help of command from the official BCFtools tutorial. A huge number of identical lines were printed. |
|
@PlatonB What command line did you use? And what was in your reference fasta file? |
I quoted part of fastq above. |
|
That's not the right fasta file. You need the one that was used to align the data and has the |
You can use |
Hello there,
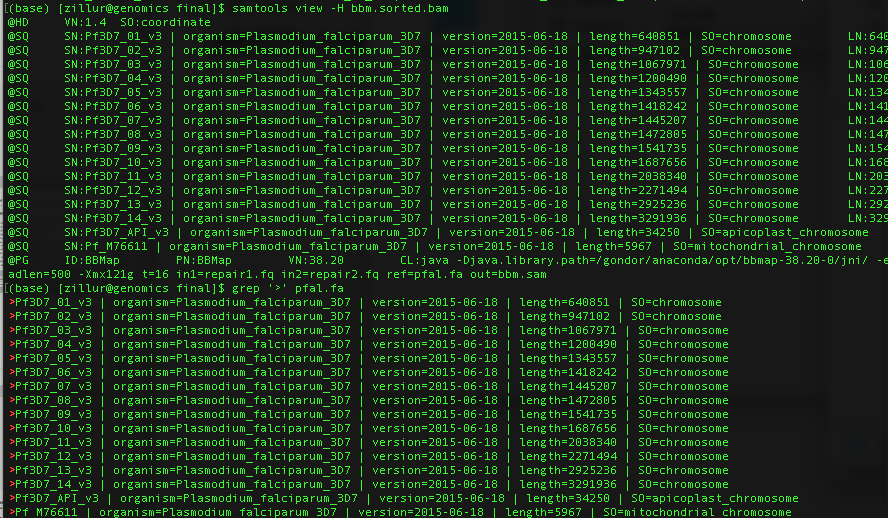
I am using samtools mpileup for snp calling. Whenever I use
samtools mpileup -uf pfal.fa bbm.sorted.bam | bcftools call -c > bbm.vcfor any mpileup command I am getting[E::faidx_adjust_position] The sequence "Pf3D7_01_v3 | organism=Plasmodium_falciparum_3D7 | version=2015-06-18 | length=640851 | SO=chromosome" not foundfor all position. I have indexed both fasta and bam file. I have tried with bcftools mpileup also but same error report. My fasta and bam headers are same (attached). Any help?Best regards
Zillur
The text was updated successfully, but these errors were encountered: